


日本には高度な医療機器を駆使するだけの高い医療技術があるにもかかわらず、機器の開発から承認までにかかる期間は、欧米に比べかなり長い。ここ数年、厚生労働省でもこの問題を解消するためさまざまな取り組みを行っており、医療機器の承認審査の合理化、効率化を図っている。現在日本が抱える審査過程の問題点と今後の方針について、行政の立場から山本弘史氏が解説した。
薬事法による医療機器の規制の仕組み
厚生労働省では医療機器の品質、有効性、安全性を確保するため、医療機器の製造業者あるいは販売業者に対して薬事法による規制を行っている。その目的は、医療上特に規制の必要性が高い医薬品および医療用具の研究開発を促進し、保健衛生の向上を図ることにある。「医療機器」について、法律では、疾病の診断、治療、予防に用いる道具で、身体の構造もしくは機能に影響を及ぼす機械器具と定められている。補助人工心臓やペースメーカー、血液透析装置、MRIやPETなどの診断装置をはじめ、コンタクトレンズや血圧計、包帯、家庭用マッサージ器など、その数は約30万種にも及ぶ。医薬品の場合、保険適用品で1万数千、いわゆる大衆薬が1万程度といわれているので、医療機器はそれよりもはるかに多い。
薬事法の改正と医療機器の安全性管理
医療機器は開発スピードが極めて速く、製品の改良もめまぐるしい。こうした特徴に対応していくため、厚生労働省では昨年4月、薬事法の改正を行った。この改正では、医療機器を国際的なリスク分類に応じて分け、X線フィルムや体温計などリスクが低いものについては規制を緩和して、ペースメーカーや人工心臓弁、血液透析装置など侵襲性の高いものに行政資源を集中して投入するという考え方で、審査の効率化や安全対策の見直しが行われた(図1)。また、リスクの高いものの販売業に許可制を導入するなど、リスクに応じた類型化を図っている。
図1. 医療機器にかかわる「カテゴリー」と「安全対策」の見直し
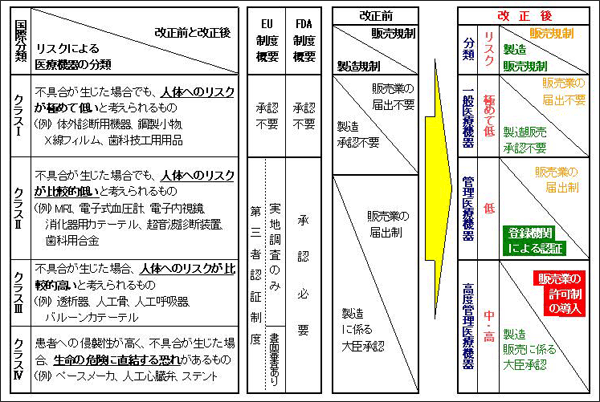
薬事法では市販後の安全管理として、メーカー側に対し、安全性情報を医療機関や国民に対して広く提供することを義務付けている。一方、行政側も、承認時あるいは承認後において、「その時点での医学的・科学的水準に照らして最も適切である対応を裁量により行うべき」と義務付けられている。山本氏は「審査の段階でも市販後の段階でも、一義的にはメーカーが責任を負うが、国としても責任を取らなければいけない仕組みになっている」と話す。
医療機器承認審査の現状
承認審査では、メーカーが提出する臨床試験成績などの資料に基づき、機器の有効性と安全性、有害性(リスク)と有用性(ベネフィット)を比較検討しながら科学的に評価し、総合判断をした上で機器を承認するかどうかが決定される。医療機器は医薬品と異なってそのメカニズムから有効性は明らかであり、より早く審査ができるのではないか、また、臨床試験は必要ないのではないかとの声もある。医学薬学上、有効性がよく知られている場合は臨床試験成績に関する資料の提出を省略することもできるが、原則として、安全性には動物実験、有効性には臨床試験、品質には物理化学試験が義務付けられている。ただ、山本氏によると、「医薬品と違って医療機器では人種差を考慮しなければならないケースが少ないため、海外の臨床試験成績をそのまま使って評価できることが多く、それらを積極的に活用し、審査の効率化を図っている」という。
一方、日本で使用されている治療関連機器の大部分は輸入に頼っているという現状がある。特許出願件数をみると、米国は日本の約7倍、ヨーロッパは3倍、研究開発費用でも、国内企業の44億円に対し、外資系企業は434億円(2001年)とその差は大きい。
新しい医療機器審査の体制
図2. 新医療機器審査の流れ
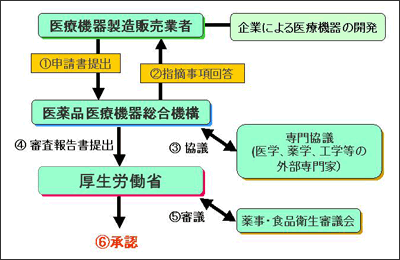
2004年、医療機器の承認審査を行う機関として、独立行政法人医薬品医療機器総合機構が設置された。これは国立医薬品食品衛生研究所と財団法人医療機器センターの行っていた審査関連業務を統合したもので、この機関の設置により新医療機器審査の体制が整えられた(図2)。 発足当時22名だった総合機構の審査官は、現在28名に増員強化され、2008年末までには35名に増員予定であるという。これに対し、米国食品医薬品局(FDA)の医療機器の審査官は314名(2004年)と圧倒的な差がある。しかし、日本でも昨年からは専門分野ごとのチーム審査を行うなど、専門性を強化し、さらに、少しでも迅速かつ高度な審査を行うため、外部の専門家にも審査を委嘱し、2005年度は1年間で延べ438名が審査に参画した。
総合機構発足前から審査が継続されていた医療機器の数も、現在では4割未満にまで減少し、今年9月までにはなくなる見込みである。また、申請企業のレベルアップのため、総合機構では事前面談や申請前、治験前相談などを実施し、厚生労働省でも企業向けの講習会を行うなどの取り組みを行っているという。山本氏は「最近では、医療機器の改良頻度の増加に対応するため、軽微な変更については届出を簡略化するなどの合理化も図っているが、さらなる検討が必要だろう」と述べ、今後への課題がまだ残っているとの認識を示した。
次世代医療機器評価指標ガイドラインの整備
新しい医療機器が世に出るまでには、基礎研究から設計・開発、安全性試験や臨床試験、そして審査・承認といった長いプロセスがある。そこで、全体のプロセスを早めるため、昨年度から次世代医療機器に対して新たな評価指標ガイドラインを整備する試験的事業が開始されている(図3)。この事業は厚生労働省の研究開発担当部門や経済産業省が協力して行っているもので、次世代医療機器に対する評価指標を早めに設定することで、企業はより効率的に開発を、行政側はより迅速に審査を進めることができるようになるという。 図3. 次世代医療機器評価指標ガイドラインの整備
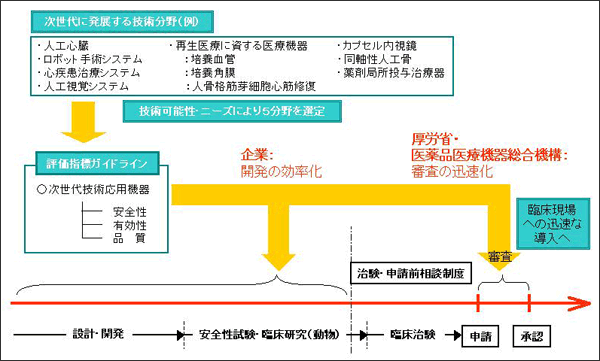
昨年は、厚生労働省、経済産業省による合同検討会が3回開催され、医療機器評価指標ガイドラインの策定対象5分野について、開発の段階からどのように審査をしていくかが検討された。合同検討会の決定はワーキンググループに通達され、事業者へのヒアリングや文献調査、実証実験、ガイドライン作成などが行われた後、再び合同検討会で審議される。 山本氏は、「こうした評価指標ガイドラインの整備への取り組みを始めてから、この1年でも前向きな成果が得られている。我々は今後も、医療機器は医薬品とは違うという特徴に着目して、設計段階から見ていくことなどで開発・審査のプロセスを迅速化できるのではないか、との考えに立ち、さらなる迅速化を目指したい」と結んだ。